ipymd.atom_analysis package¶
Subpackages¶
Submodules¶
ipymd.atom_analysis.basic module¶
Created on Thu Jul 14 14:06:40 2016
@author: cjs14
functions to calculate basic properties of the atoms
-
ipymd.atom_analysis.basic.density_bb(atoms_df, vectors=[[1, 0, 0], [0, 1, 0], [0, 0, 1]])[source]¶ calculate density of the bounding box (assuming all atoms are inside)
-
ipymd.atom_analysis.basic.lattparams_bb(vectors=[[1, 0, 0], [0, 1, 0], [0, 0, 1]], rounded=None, cells=(1, 1, 1))[source]¶ calculate unit cell parameters of the bounding box
- rounded : int
- the number of decimal places to return
- cells : (int,int,int)
- how many unit cells the vectors represent in each direction
Returns: Return type: a, b, c, alpha, beta, gamma (in degrees)
-
ipymd.atom_analysis.basic.volume_bb(vectors=[[1, 0, 0], [0, 1, 0], [0, 0, 1]], rounded=None, cells=(1, 1, 1))[source]¶ calculate volume of the bounding box
- rounded : int
- the number of decimal places to return
- cells : (int,int,int)
- how many unit cells the vectors represent in each direction
Returns: volume Return type: float
ipymd.atom_analysis.nearest_neighbour module¶
Created on Thu Jul 14 14:05:09 2016
@author: cjs14
functions based on nearest neighbour calculations
-
ipymd.atom_analysis.nearest_neighbour.bond_lengths(atoms_df, coord_type, lattice_type, max_dist=4, max_coord=16, repeat_meta=None, rounded=2, min_dist=0.01, leafsize=100)[source]¶ calculate the unique bond lengths atoms in coords_atoms, w.r.t lattice_atoms
- atoms_df : pandas.Dataframe
- all atoms
- coord_type : string
- atoms to calcualte coordination of
- lattice_type : string
- atoms to act as lattice for coordination
- max_dist : float
- maximum distance for coordination consideration
- max_coord : float
- maximum possible coordination number
- repeat_meta : pandas.Series
- include consideration of repeating boundary idenfined by a,b,c in the meta data
- min_dist : float
- lattice points within this distance of the atom will be ignored (assumed self-interaction)
- leafsize : int
- points at which the algorithm switches to brute-force (kdtree specific)
Returns: distances – list of unique distances Return type: set
-
ipymd.atom_analysis.nearest_neighbour.cna_categories(atoms_df, accuracy=1.0, upper_bound=4, max_neighbours=24, repeat_meta=None, leafsize=100, ipython_progress=False)[source]¶ compute summed atomic environments of each atom in atoms_df
Based on Faken, Daniel and Jónsson, Hannes, ‘Systematic analysis of local atomic structure combined with 3D computer graphics’, March 1994, DOI: 10.1016/0927-0256(94)90109-0
signatures: - FCC = 12 x 4,2,1 - HCP = 6 x 4,2,1 & 6 x 4,2,2 - BCC = 6 x 6,6,6 & 8 x 4,4,4 - Diamond = 12 x 5,4,3 & 4 x 6,6,3 - Icosahedral = 12 x 5,5,5
Parameters: - accuracy (float) – 0 to 1 how accurate to fit to signature
- repeat_meta (pandas.Series) – include consideration of repeating boundary idenfined by a,b,c in the meta data
- ipython_progress (bool) – print progress to IPython Notebook
Returns: df – copy of atoms_df with new column named cna
Return type: pandas.Dataframe
-
ipymd.atom_analysis.nearest_neighbour.cna_plot(atoms_df, upper_bound=4, max_neighbours=24, repeat_meta=None, leafsize=100, barwidth=1, ipython_progress=False)[source]¶ compute summed atomic environments of each atom in atoms_df
Based on Faken, Daniel and Jónsson, Hannes, ‘Systematic analysis of local atomic structure combined with 3D computer graphics’, March 1994, DOI: 10.1016/0927-0256(94)90109-0
common signatures: - FCC = 12 x 4,2,1 - HCP = 6 x 4,2,1 & 6 x 4,2,2 - BCC = 6 x 6,6,6 & 8 x 4,4,4 - Diamond = 12 x 5,4,3 & 4 x 6,6,3 - Icosahedral = 12 x 5,5,5
Parameters: - repeat_meta (pandas.Series) – include consideration of repeating boundary idenfined by a,b,c in the meta data
- ipython_progress (bool) – print progress to IPython Notebook
Returns: plot – a matplotlib plot
Return type:
-
ipymd.atom_analysis.nearest_neighbour.cna_sum(atoms_df, upper_bound=4, max_neighbours=24, repeat_meta=None, leafsize=100, ipython_progress=False)[source]¶ compute summed atomic environments of each atom in atoms_df
Based on Faken, Daniel and Jónsson, Hannes, ‘Systematic analysis of local atomic structure combined with 3D computer graphics’, March 1994, DOI: 10.1016/0927-0256(94)90109-0
common signatures: - FCC = 12 x 4,2,1 - HCP = 6 x 4,2,1 & 6 x 4,2,2 - BCC = 6 x 6,6,6 & 8 x 4,4,4 - Diamond = 12 x 5,4,3 & 4 x 6,6,3 - Icosahedral = 12 x 5,5,5
Parameters: - repeat_meta (pandas.Series) – include consideration of repeating boundary idenfined by a,b,c in the meta data
- ipython_progress (bool) – print progress to IPython Notebook
Returns: counter – a counter of cna signatures
Return type: Counter
-
ipymd.atom_analysis.nearest_neighbour.common_neighbour_analysis(atoms_df, upper_bound=4, max_neighbours=24, repeat_meta=None, leafsize=100, ipython_progress=False)[source]¶ compute atomic environment of each atom in atoms_df
Based on Faken, Daniel and Jónsson, Hannes, ‘Systematic analysis of local atomic structure combined with 3D computer graphics’, March 1994, DOI: 10.1016/0927-0256(94)90109-0
ideally: - FCC = 12 x 4,2,1 - HCP = 6 x 4,2,1 & 6 x 4,2,2 - BCC = 6 x 6,6,6 & 8 x 4,4,4 - icosahedral = 12 x 5,5,5
- repeat_meta : pandas.Series
- include consideration of repeating boundary idenfined by a,b,c in the meta data
- ipython_progress : bool
- print progress to IPython Notebook
Returns: df – copy of atoms_df with new column named cna Return type: pandas.Dataframe
-
ipymd.atom_analysis.nearest_neighbour.compare_to_lattice(atoms_df, lattice_atoms_df, max_dist=10, leafsize=100)[source]¶ calculate the minimum distance of each atom in atoms_df from a lattice point in lattice_atoms_df
- atoms_df : pandas.Dataframe
- atoms to calculate for
- lattice_atoms_df : pandas.Dataframe
- atoms to act as lattice points
- max_dist : float
- maximum distance for consideration in computation
- leafsize : int
- points at which the algorithm switches to brute-force (kdtree specific)
Returns: distances – list of distances to nearest atom in lattice Return type: list
-
ipymd.atom_analysis.nearest_neighbour.coordination(coord_atoms_df, lattice_atoms_df, max_dist=4, max_coord=16, repeat_meta=None, min_dist=0.01, leafsize=100)[source]¶ calculate the coordination number of each atom in coords_atoms, w.r.t lattice_atoms
- coords_atoms_df : pandas.Dataframe
- atoms to calcualte coordination of
- lattice_atoms_df : pandas.Dataframe
- atoms to act as lattice for coordination
- max_dist : float
- maximum distance for coordination consideration
- max_coord : float
- maximum possible coordination number
- repeat_meta : pandas.Series
- include consideration of repeating boundary idenfined by a,b,c in the meta data
- min_dist : float
- lattice points within this distance of the atom will be ignored (assumed self-interaction)
- leafsize : int
- points at which the algorithm switches to brute-force (kdtree specific)
Returns: coords – list of coordination numbers Return type: list
-
ipymd.atom_analysis.nearest_neighbour.coordination_bytype(atoms_df, coord_type, lattice_type, max_dist=4, max_coord=16, repeat_meta=None, min_dist=0.01, leafsize=100)[source]¶ returns dataframe with additional column for the coordination number of each atom of coord type, w.r.t lattice_type atoms
effectively an extension of calc_df_coordination
- atoms_df : pandas.Dataframe
- all atoms
- coord_type : string
- atoms to calcualte coordination of
- lattice_type : string
- atoms to act as lattice for coordination
- max_dist : float
- maximum distance for coordination consideration
- max_coord : float
- maximum possible coordination number
- repeat_meta : pandas.Series
- include consideration of repeating boundary idenfined by a,b,c in the meta data
- min_dist : float
- lattice points within this distance of the atom will be ignored (assumed self-interaction)
- leafsize : int
- points at which the algorithm switches to brute-force (kdtree specific)
Returns: df – copy of atoms_df with new column named coord_{coord_type}_{lattice_type} Return type: pandas.Dataframe
-
ipymd.atom_analysis.nearest_neighbour.guess_bonds(atoms_df, covalent_radii=None, threshold=0.1, max_length=5.0, radius=0.1, transparency=1.0, color=None)[source]¶ guess bonds between atoms, based on approximate covalent radii
Parameters: - atoms_df (pandas.Dataframe) – all atoms, requires colums [‘x’,’y’,’z’,’type’, ‘color’]
- covalent_radii (dict or None) – a dict of covalent radii for each atom type, if None then taken from ipymd.shared.atom_data
- threshold (float) – include bonds with distance +/- threshold of guessed bond length (Angstrom)
- max_length (float) – maximum bond length to include (Angstrom)
- radius (float) – radius of displayed bond cylinder (Angstrom)
- transparency (float) – transparency of displayed bond cylinder
- color (str or tuple) – color of displayed bond cylinder, if None then colored by atom color
Returns: bonds_df – a dataframe with start/end indexes relating to atoms in atoms_df
Return type: pandas.Dataframe
-
ipymd.atom_analysis.nearest_neighbour.vacancy_identification(atoms_df, res=0.2, nn_dist=2.0, repeat_meta=None, remove_dups=True, color='red', transparency=1.0, radius=1, type_name='Vac', leafsize=100, n_jobs=1, ipython_progress=False)[source]¶ identify vacancies
- atoms_df : pandas.Dataframe
- atoms to calculate for
- res : float
- resolution of vacancy identification, i.e. spacing of reference lattice
- nn_dist : float
- maximum nearest-neighbour distance considered as a vacancy
- repeat_meta : pandas.Series
- include consideration of repeating boundary idenfined by a,b,c in the meta data
- remove_dups : bool
- only keep one vacancy site within the nearest-neighbour distance
- leafsize : int
- points at which the algorithm switches to brute-force (kdtree specific)
- n_jobs : int, optional
- Number of jobs to schedule for parallel processing. If -1 is given all processors are used.
- ipython_progress : bool
- print progress to IPython Notebook
Returns: vac_df – new atom dataframe of vacancy sites as atoms Return type: pandas.DataFrame
ipymd.atom_analysis.spectral module¶
Created on Tue Jul 12 20:18:04 2016
Derived from: LAMMPS - Large-scale Atomic/Molecular Massively Parallel Simulator http://lammps.sandia.gov, Sandia National Laboratories Steve Plimpton, sjplimp@sandia.gov Copyright (2003) Sandia Corporation. Under the terms of Contract DE-AC04-94AL85000 with Sandia Corporation, the U.S. Government retains certain rights in this software. This software is distributed under the GNU General Public License. https://github.com/lammps/lammps/tree/lammps-icms/src/USER-DIFFRACTION
This package contains the commands neeed to calculate x-ray and electron diffraction intensities based on kinematic diffraction theory. Detailed discription of the computation can be found in the following works:
Coleman, S.P., Spearot, D.E., Capolungo, L. (2013) Virtual diffraction analysis of Ni [010] symmetric tilt grain boundaries, Modelling and Simulation in Materials Science and Engineering, 21 055020. doi:10.1088/0965-0393/21/5/055020
Coleman, S.P., Sichani, M.M., Spearot, D.E. (2014) A computational algorithm to produce virtual x-ray and electron diffraction patterns from atomistic simulations, JOM, 66 (3), 408-416. doi:10.1007/s11837-013-0829-3
Coleman, S.P., Pamidighantam, S. Van Moer, M., Wang, Y., Koesterke, L. Spearot D.E (2014) Performance improvement and workflow development of virtual diffraction calculations, XSEDE14, doi:10.1145/2616498.2616552
@author: chris sewell
-
ipymd.atom_analysis.spectral.xrd_compute(atoms_df, meta_data, wlambda, min2theta=1.0, max2theta=179.0, lp=True, rspace=[1, 1, 1], manual=False, periodic=[True, True, True])[source]¶ Compute predicted x-ray diffraction intensities for a given wavelength
- atoms_df : pandas.DataFrame
- a dataframe of info for each atom, including columns; x,y,z,type
- meta_data : pandas.Series
- data of a,b,c crystal vectors (as tuples, e.g. meta_data.a = (0,0,1))
- wlambda : float
- radiation wavelength (length units) typical values are Cu Ka = 1.54, Mo Ka = 0.71 Angstroms
- min2theta : float
- minimum 2 theta range to explore (degrees)
- max2theta : float
- maximum 2 theta range to explore (degrees)
- lp : bool
- switch to apply Lorentz-polarization factor
- use_triclinic : bool
- use_triclinic
- rspace : list of floats
- parameters to adjust the spacing of the reciprocal lattice nodes in the h, k, and l directions respectively
- manual : bool
- use manual spacing of reciprocal lattice points based on the values of the c parameters (good for comparing diffraction results from multiple simulations, but small c required).
- periodic : list of bools
- whether periodic boundary in the h, k, and l directions respectively
Returns: df – theta (in degrees), I, h, k and l for each k-point Return type: pandas.DataFrame Notes
This is an implementation of the virtual x-ray diffraction pattern algorithm by Coleman et al. [ref1]
The algorithm proceeds in the following manner:
- Define a crystal structure by position (x,y,z) and atom/ion type.
- Define the x-ray wavelength to use
- Compute the full reciprocal lattice mesh
- Filter reciprocal lattice points by those in the Eswald’s sphere
- Compute the structure factor at each reciprocal lattice point, for each atom type
- Compute the x-ray diffraction intensity at each reciprocal lattice point
- Group and sum intensities by angle
reciprocal points of the lattice are computed such that:
\[\mathbf{K} = {m_{1}}\cdot \mathbf{b}_{1}+{m_{2}}\cdot \mathbf{b}_{2}+{m_{3}}\cdot \mathbf{b}_{3}\]where,
\[\begin{split}\begin{aligned} \mathbf {b_{1}} &= {\frac {\mathbf {a_{2}} \times \mathbf {a_{3}} }{\mathbf {a_{1}} \cdot (\mathbf {a_{2}} \times \mathbf {a_{3}} )}}\\ \mathbf {b_{2}} &= {\frac {\mathbf {a_{3}} \times \mathbf {a_{1}} }{\mathbf {a_{2}} \cdot (\mathbf {a_{3}} \times \mathbf {a_{1}} )}}\\ \mathbf {b_{3}} &= {\frac {\mathbf {a_{1}} \times \mathbf {a_{2}} }{\mathbf {a_{3}} \cdot (\mathbf {a_{1}} \times \mathbf {a_{2}} )}} \end{aligned}\end{split}\]The reciprocal k-point modulii of the x-ray is calculated from Bragg’s law:
\[\left| {\mathbf{K}_{\lambda}} \right| = \frac{1}{{d_{\text{hkl}} }} = \frac{2\sin \left( \theta \right)}{\lambda }\]This is used to construct an Eswald’s sphere, and only reciprocal lattice ponts within are retained, as illustrated:
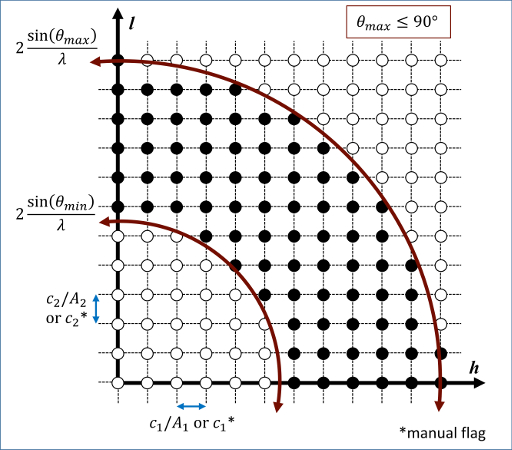
The atomic scattering factors, fj, accounts for the reduction in diffraction intensity due to Compton scattering, with coefficients based on the electron density around the atomic nuclei.
\[f_j \left(\frac{\sin \theta}{\lambda}\right) = \left[ \sum\limits^4_i a_i \exp \left( -b_i \frac{\sin^2 \theta}{\lambda^2} \right)\right] + c = \left[ \sum\limits^4_i a_i \exp \left( -b_i \left(\frac{\left| {\mathbf{K}} \right|}{2}\right)^2 \right)\right] + c\]The relative diffraction intensity from x-rays is computed at each reciprocal lattice point through:
\[I_x(\mathbf{K}) = Lp(\theta) \frac{F(\mathbf{K})F^*(\mathbf{K})}{N}\]such that:
\[F ({\mathbf{K}} )= \sum\limits_{j = 1}^{N} {f_{j}.e^{\left( {2\pi i \, {\mathbf{K}} \cdot {\mathbf{r}}_{j} } \right)}} = \sum\limits_{j = 1}^{N} {f_j.\left[ \cos \left( 2\pi \mathbf{K} \cdot \mathbf{r}_j \right) + i \sin \left( 2\pi \mathbf{K} \cdot \mathbf{r}_j \right) \right]}\]and the Lorentz-polarization factor is:
\[Lp(\theta) = \frac{1+\cos^2 (2\theta)}{\cos(\theta)\sin^2(\theta)}\]References
[ref1] 1.Coleman, S. P., Sichani, M. M. & Spearot, D. E. A Computational Algorithm to Produce Virtual X-ray and Electron Diffraction Patterns from Atomistic Simulations. JOM 66, 408–416 (2014).
-
ipymd.atom_analysis.spectral.xrd_group_i(df, tstep=None, sym_equiv_hkl='none')[source]¶ group xrd intensities by theta
Parameters: - df (pandas.DataFrame) – containing columns; [‘theta’,’I’] and optional [‘h’,’k’,’l’]
- tstep (None or float) – if not None, group thetas within ranges i to i+tstep
- sym_equiv_hkl (str) – group hkl by symmetry-equivalent refelections; [‘none’,’mmm’,’m-3m’]
Returns: df
Return type: Notes
if grouping by theta step, then the theta value for each group will be the intensity weighted average
Crystal System | Laue Class | Symmetry-Equivalent Reflections | Multiplicity ————– | ———- | ——————————- | ———— Triclinic | -1 | hkl ≡ -h-k-l | 2 Monoclinic | 2/m | hkl ≡ -hk-l ≡ -h-k-l ≡ h-kl | 4 Orthorhombic | mmm | hkl ≡ h-k-l ≡ -hk-l ≡ -h-kl ≡ -h-k-l ≡ -hkl ≡ h-kl ≡ hk-l | 8 Tetragonal | 4/m | hkl ≡ -khl ≡ -h-kl ≡ k-hl ≡ -h-k-l ≡ k-h-l ≡ hk-l ≡ -kh-l | 8
4/mmm | hkl ≡ -khl ≡ -h-kl ≡ k-hl ≡ -h-k-l ≡ k-h-l ≡ hk-l ≡ -kh-l || ≡ khl ≡ -hkl ≡ -k-hl ≡ h-kl ≡ -k-h-l ≡ h-k-l ≡ kh-l ≡ -hk-l | 16- Cubic | m-3 | hkl ≡ -hkl ≡ h-kl ≡ hk-l ≡ -h-k-l ≡ h-k-l ≡ -hk-l ≡ -h-kl |
- | ≡ klh ≡ -klh ≡ k-lh ≡ kl-h ≡ -k-l-h ≡ k-l-h ≡ -kl-h ≡ -k-lh || ≡ lhk ≡ -lhk ≡ l-hk ≡ lh-k ≡ -l-h-k ≡ l-h-k ≡ -lh-k ≡ -l-hk | 24m-3m | hkl ≡ -hkl ≡ h-kl ≡ hk-l ≡ -h-k-l ≡ h-k-l ≡ -hk-l ≡ -h-kl || ≡ klh ≡ -klh ≡ k-lh ≡ kl-h ≡ -k-l-h ≡ k-l-h ≡ -kl-h ≡ -k-lh || ≡ lhk ≡ -lhk ≡ l-hk ≡ lh-k ≡ -l-h-k ≡ l-h-k ≡ -lh-k ≡ -l-hk || ≡ khl ≡ -khl ≡ k-hl ≡ kh-l ≡ -k-h-l ≡ k-h-l ≡ -kh-l ≡ -k-hl || ≡ lkh ≡ -lkh ≡ l-kh ≡ lk-h ≡ -l-k-h ≡ l-k-h ≡ -lk-h ≡ -l-kh || ≡ hlk ≡ -hlk ≡ h-lk ≡ hl-k ≡ -h-l-k ≡ h-l-k ≡ -hl-k ≡ -h-lk | 48
-
ipymd.atom_analysis.spectral.xrd_plot(df, icol='I_norm', imin=0.01, barwidth=1.0, hkl_num=0, hkl_trange=[0.0, 180.0], incl_multi=False, label_inline=True, label_trange=[0.0, 180.0], ax=None, **kwargs)[source]¶ create plot of xrd pattern
- df : pd.DataFrame
- containing columns [‘theta’,icol] and optional [‘hkl’,’multiplicity’]
- icol : str
- column name for intensity data
- imin : float
- minimum intensity to display
- barwidth : float or None
- if None then the barwidth will be the bin width
- hkl_num : int
- number of k-point values to label
- hkl_trange : [float,float]
- theta range within which to label peaks with k-point values
- label_inline : bool
- place k-point labels inline or at top of plot
- label_trange : [float,float]
- if not inline, theta range within which to fit k-point labels
- incl_multi : bool
- add multiplicity to k-point label
- kwargs : optional
- additional arguments for bar plot (e.g. label, color, alpha)
Returns: plot – a plot object Return type: ipymd.plotting.Plotting